c******r
发帖数: 3778 | 1 明显4度的和室温的发生了降解。细条带是降解产物。应该还有条相应的小分子条带,
可能EB跑出去了,所以看不清楚了。maxi没做干净,还有enzyme的contamination,所
以这个降解是比较特意性的,而不是smear。
室温的降解更明显,full length的已经全部降解,但是那个细条带由于继续水解的速
度远不如从full length降解的速度,所以还比较明显。但是多放几天,这个条带也会
消失。
艳阳天说了,要加EDTA才能防止水解,但是有时候EDTA会影响下面的反应,最好还是-
20度或者-80度保存比较安全方便。
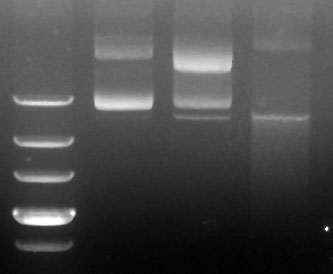
没有切过。最左边的是ladder,然后从左到右依次是冻在-20度的,藏在4度的和丢在室
温的。过了五天拿出来电泳,看看降解得怎么样了。如大家所见,最左边的条带(-20
度)看上去挺好,超螺旋和开环的分明。但是中间和右边的已经降解了,在超螺旋条带
下面又多出一条细细的但是明显的小条带。请问谁知道这个小条带是怎么回事呢? |
|
e***o
发帖数: 344 | 2 最近跑DNA胶很邪门,首先是基因组DNA的条带大小在不同的胶里跑得不一样。做3C-
library,所以在提取的过程中没有除去蛋白质,用裂解液裂解细胞后,离心收集细胞
核,再分别用SDS和Triton X-100裂解细胞核后跑的琼脂糖胶。
其中2,3泳道是刚提取完时跑的电泳条带,1,4孔道是将同样的样品放置6个小时左右
后跑的电泳条带,结果条带大小就发生了改变,有15000bp左右减小到了几百bp,而且
每次跑胶,电泳条带的大小都不固定,大多都只有几百bp,和基因组大小不符!
然后是回收cDNA500-700bp的带加adapter,扩PCR也出现这种飘移现象。到别的lab也试
了,还是这样。跑别的特异PCR扩增和酶切质粒都是好好地。真是邪门了。
请大家分析下是什么原因造成的。谢谢! |
|
m****m
发帖数: 395 | 3 最近提纯一个HIS-TAG的蛋白,试了很久就是不是一条带,一直以为是杂带没有清除,
后来感觉不对劲,好像就是它本身。但是很奇怪,如果是这个蛋白的二聚体、三聚体等
,应该是跑在整数倍的线上啊,但是不是,可这些条带又是线性有规则地排开的。还有
,有时候同一条带上的一条蛋白会分裂,就是靠得非常近,但是仔细看是分裂的。有谁
能帮我解释一下为什么会跑出这种胶的结果来吗?同一个蛋白构象不同,条带也会不在
一个地方吗? |
|
m***o
发帖数: 272 | 4 刚开始去一个lab,做PCR,最近发现阴性对照组经常有跟目的片段一样大小的条带。用
过不同primer,那个片段就跟我的目的片段一样变。如果是水污染了,但是有些管PCR
就什么都没有。因为做的是5和3’RACE,有时候第一次PCR阴性对照组很干净,在用这
个做nest PCR就有了。感觉如果cycle多用的话,阴性对照组就会出来条带。老板今天
专门强调这是技术问题,还是绝对的技术问题。郁闷的不行,唉,刚开始来这个lab,
感觉这个实验室挺脏,是不是环境不好,但是有些组就什么条带没有。但是阴性对照是
经常有,而且条带亮的快赶上阳性对照了。各位,可能是什么问题呢?谢谢任何建议! |
|
t*****z
发帖数: 1598 | 5 图在这里,看不到的点击链接啊:
[img]http://www2.picturepush.com/photo/a/6117040/img/6117040.jpg[/img]
我在琢磨大质粒的降解和构象变化的问题。这是一个大约17kb的质粒,大抽出来的,没有切过。最左边的是ladder,然后从左到右依次是冻在-20度的,藏在4度的和丢在室温的。过了五天拿出来电泳,看看降解得怎么样了。如大家所见,最左边的条带(-20度)看上去挺好,超螺旋和开环的分明。但是中间和右边的已经降解了,在超螺旋条带下面又多出一条细细的但是明显的小条带。请问谁知道这个小条带是怎么回事呢?
多谢啦! |
|
g*****n
发帖数: 241 | 6 我FLAG IP了一个蛋白,然后跑western,用anti FLAG得到blot之后strip membrane,
然后用抗蛋白本身的抗体再probe一遍,结果发现两次得到的条带不重叠 (相差不远,
但很明显不重叠)。FLAG抗体出现两条带,高的带明显,低的带比较浅。但抗蛋白本身
的抗体得到一条带,和低的那条带差不多重叠。
本来是想用两个抗体各检测一遍更严谨点,没想到出了这样的结果反而麻烦了。请问大
家这是怎么回事。
谢谢 |
|
C*******4
发帖数: 400 | 7 首先排除抗体本身的中轻链(~50和25kD), 其次Flag在~35kDa左右能拉和反应对一条
非特异带。很常见。要甄别一下。
[在 geosmin (Little+Monkey) 的大作中提到:]
:我FLAG IP了一个蛋白,然后跑western,用anti FLAG得到blot之后strip membrane,
:然后用抗蛋白本身的抗体再probe一遍,结果发现两次得到的条带不重叠 (相差不远
,但很明显不重叠)。FLAG抗体出现两条带,高的带明显,低的带比较浅。但抗蛋白本
身的抗体得到一条带,和低的那条带差不多重叠。
:........... |
|
C*******4
发帖数: 400 | 8 可能这个蛋白不稳定,一部分有降解,但FLAG还都在。抗其自己的抗体因为要检测的那
一段缺失了,就不能检测到。试一试抗另一边的抗体。如原来的是抗N端序列,换一个C
端的试一试。
[在 geosmin (Little+Monkey) 的大作中提到:]
:我FLAG IP了一个蛋白,然后跑western,用anti FLAG得到blot之后strip membrane,
:然后用抗蛋白本身的抗体再probe一遍,结果发现两次得到的条带不重叠 (相差不远
,但很明显不重叠)。FLAG抗体出现两条带,高的带明显,低的带比较浅。但抗蛋白本
身的抗体得到一条带,和低的那条带差不多重叠。
:........... |
|
|
v***a
发帖数: 1242 | 9 要克隆一个gene,发现用我自己培养的细胞来PCR扩增时,大部分P不出,少数P得出。
用别人养的相同的细胞,都P不出。凡是P出来的,会有两条靠得很近很近的条带(~1.
5kb)。
然后我就挖了上面的那一条下来,做成plasmid了,测序也都完全正确。
这次要做recombinant protein,所以重新设计了primer(PCR产物很短,只有600多bp
),用上次做的现成的plasmid进行PCR,结果又P出来两条很近的条带。
有点想不通,请教一下大家这是怎么回事呢?
另,做western blot时偶尔也出现靠得很近的条带(忽而又只有一条),没有见过有相
关isoform的报导。
谢谢! |
|
G***G
发帖数: 16778 | 10 邻居割完草,会有条带纹。但是我的不是很明显。
请问大家有经验的,如何弄出深浅不一的条带纹?
需要在割草机上设置什么吗? |
|
m****m
发帖数: 395 | 11 要提纯蛋白,可以看到提纯前虽然有些杂带,但是目标蛋白还是一条带在那里的,于是
我用NI柱提纯,其中用0.1%SDS洗柱子去杂蛋白,最后用1%SDS洗脱蛋白,洗柱IMIDAZO
浓度36mM,洗脱IMIDAZO浓度500mM,取样都用BME切了(5%BME,冷室overnight),结果
用纯化前蛋白、洗柱取样、纯化后的蛋白跑胶完后发现,纯化前的目标蛋白还是一条带
(有杂质),但是经过SDS洗和洗脱后的样品都发现我要的蛋白居然分裂成两条临近的
带,距离有5KD左右,都挺深的(一条稍微浅点),太奇怪了,想不通! |
|
m****m
发帖数: 395 | 12 有没有有经验的啊?
1ul的样品,加1*SB至20ul后全部加入胶孔,最后条带亮度中等,也就是,
marker上了3ul,比marker的那个条带要亮1倍左右。
估计我这1ul的样品里有多少ug蛋白呢?
样品总共700ul,我想知道这700ul里有多少mg的蛋白。 |
|
R*****w
发帖数: 447 | 13 需要你自己设计序列,让公司合成的。这段tail序列可以是任意序列,但你需要先
blast一下,不要找个与human或mouse的genome相似性很高的,否则你会有麻烦。
理论上会有很多条带,甚至smear,但实际上只会有1-3条带,你可以只挑最大的那条
带做克隆测序就可以了。
good luck |
|
c***y
发帖数: 615 | 14 单引物产物应该不能形成DNA双螺旋的,怎么可能在EB胶上检测到闪亮的条带?原模板
是cDNA pool,扩增之前检测不到这条带,几乎就是均匀分布的smear。 |
|
g******u
发帖数: 66 | 15 western blot的条带大小是一致的.弱的表达就是条带的density在knockout动物组织中
低于wildtype动物组织.
您说的对,发表的WB图是很容易把弱的background去除掉.但这个事实还是存在,欺骗不
了自己啊. |
|
h**********r
发帖数: 671 | 16 定量SDS-PAGE表达的条带有什么free的软件啊?就是想看看特定的条带占总量的百分比
之类的。多谢! |
|
g*****n
发帖数: 241 | 17 谢谢大家的回复,我试试把曝光时间弄长点看能不能看到两条带。有可能是PTM导致了
其中一条带不容易被我的抗体识别。
可惜我们只有一个自己的抗体,否则可以按照culater94的建议试试两端不同的。 |
|
m****m
发帖数: 395 | 18 这次加了BME了,还是有这个现象,明明是同一个蛋白,在不同泳道上,却跑得一前一
后,虽然间隔极近,几乎就快要一直线了,但是还是有明显的前后。我用FPLC分离的,
看曲线分段取样,然后跑在不同的道上,其实肯定是同一个东西,条带很深,杂蛋白不
带HIS-TAG,之前WASH步骤应该就洗掉了,不可能结合得那么牢,最后ELUTE的时候下来
一大堆,肯定是样品,但是不知道为什么还是这样。或者跟上样量也会有关吗?比如上
得多会跑得慢点?
我是提纯了用来测结构的,所以希望尽可能纯。 |
|
e****p
发帖数: 354 | 19 做3’RACE 快半年了,用了好几种TAQ - strategen pfu, takara, biorad bioline,
invitrogen platium hifi. 3‘引物设计了6个,条件也优化(AT,elonging time, mg+
),要不什么也没有要不没有P出目的条带(测序后错配了)。。。唉 都要崩溃了..
另外的一个基因到是P出来了,这个基因3'端有很多重复序 GC 50-60%的样子 请教大家
一般用什么办法或强大的酶。。。
非常感谢 |
|
e****p
发帖数: 354 | 20 请问这个随机引物是自己合成的还是可以直接买到的啊?
这样的话RT后 PCR会有很多条带吧,
谢谢!
“5 |
|
m***o
发帖数: 272 | 21 看来老板没冤枉我吗?我是察了,但是如果就一轮PCR 看起来都还好,但是在继续nest
PCR 一轮就很容易出来了,明天我把试验台也察察看。为什么有些样品里面我怎么都
做不出条带,而这样阴性对照就容易出来。而且我现在都是先加阴性对照组的水,盖上
盖子,然后再做别的和阳性对照。我真不知道操作该怎么改进。或者我不该做阳性对照
组,可能都是它惹的。 |
|
b******n
发帖数: 4225 | 22 denatured plasmid which is resistant to enzymatical digestion
it's due to long time treatment of solution II(NaOH/SDS)
没有切过。最左边的是ladder,然后从左到右依次是冻在-20度的,藏在4度的和丢在室
温的。过了五天拿出来电泳,看看降解得怎么样了。如大家所见,最左边的条带(-20
度)看上去挺好,超螺旋 |
|
t*****z
发帖数: 1598 | 23 是这样啊。可是我刚刚抽提好的质粒,是没有这一条带的,放上几天才出现。是否放置
本身也会让质粒变性呢?
20 |
|
g******u
发帖数: 66 | 24 最近被一个问题困扰着:
用某种通道蛋白的抗体(通过Western blot和immunohistochemistry) 来研究此蛋白在
一个组织中的表达情况.在大批量的实验前,先做了个预实验测试了下此抗体的特异性.
很奇怪的是,Western blot和immunohistochemistry 都显示在此蛋白完全敲除的动物组
织中还是有弱的阳性表达(和wild-type相比是很弱,但还是能看到非常弱的条带或者染
色).
难道是一抗或者二抗的浓度过大?还是封闭的不够?
注: 1. 此抗体已经有几篇发表的文章显示是特异性的
2. 基因敲除的动物应该没有问题
3. 弱的阳性表达应该不是来自自发荧光或者artifact
非常感谢您的建议和分析!!! |
|
g******u
发帖数: 66 | 25 谢谢!没打算PS,这个技巧可不敢玩!
就是有点纳闷为什么已经发表的文章中KO动物的组织中连很弱很弱的条带都没有,完全
是干净的!!! |
|
T*****n
发帖数: 897 | 26 作了一个PCR,本来是预期1000bp的带,但却出现了两条550和750的两条带。这是怎么
回事?还有另外两个片段,用正常捐血者的标本能搞得到预期的带,但开始测病人的样
本,却没有出现任何带。这是怎么回事?谢谢! |
|
c**a
发帖数: 94 | 27 那为什么有些sample有,有些sample里没有呢. 而且blot for total JNK的时候就没有
那条带 |
|
D**F
发帖数: 54 | 28 最近用PCR鉴定 loxp小鼠
在flox区域两侧设计引物, 理论上WT 产物较短,floxed产物较长
但奇怪的是,要么只看到长的产物,要么只看到短的产物,从来没有见到过同时有长短
两条带
根据交配后代的结果显然那些只看到 长产物的小鼠中是有 loxp +/- 杂合体的
有人遇到过这种情况吗?
感觉是floxed allele在PCR中抑制了WT的扩增
但为什么会造成这种抑制实在想不明白 |
|
t******y
发帖数: 716 | 29 应该是蛋白形成二聚体,外源蛋白和内源蛋白一起ip下来,flag的外源蛋白分子量大在
上面,内源蛋白本身在下面。蛋白的抗体可能识别的位点是和flag相连的末端,因为连
上flag反而不能识别外源蛋白了,所以只看到低一点的条带。 |
|
e****s
发帖数: 1125 | 30 这个蛋白有PTM? 如果这个Modification 正好在抗体的识别Epitope 上或者附近,会影
响抗体的识别吧?
把蛋白本身抗体曝光长点,说不定也能看见两条带?这个我倒遇见过,就是抗体对同一
蛋白不同状态有不同的敏感性。 |
|
m**********2
发帖数: 6568 | 31 还有个终极办法就是把两条带都切了mass spec |
|
发帖数: 1 | 32 韩春雨论文的实验结果是不是伪造的?
·方舟子·
河北科大副教授(据闻已突击评为正教授)韩春雨因发明基因编辑新技术,
被称为“诺贝尔级成果”,一夜成名,号称“诺公招手”了(注)。但是有多家
实验室反映重复不出其论文中最关键的图4结果。有人仔细研究图4图片,发现其
电泳结果不合理;还有人对这些图片做了亮度、对比度处理,发现有拼接的痕迹。
这是怎么回事呢?
我先给不是生物医学领域的读者粗略介绍一下其中的背景知识。论文图4是
对DNA片段做电泳的结果。DNA片段带有电荷,把它添加到凝胶中,放进缓冲液,
通上电,DNA片段就能在凝胶中移动,片段大的移动得慢,片段小的移动得快,
染色以后可以看出这些片段在哪里,再跟已知大小的标识片段做对比,就可以知
道片段的大小了。这就是所谓电泳。
论文图4a是对DNA进行了编辑后产生的不同片段进行电泳的结果。DNA是由一
个个核苷酸链接而成的,核苷酸越多,DNA片段就越大。经过剪切之后,图中的
G6和G13相差30个核苷酸。这个大小差别是不是能在电泳上看出来,取决于凝胶
的浓度和电泳的时... 阅读全帖 |
|
U*******n
发帖数: 824 | 33 在导师任先达的指导下完成学位论文一个月后,李红良迎来了答辩。这也意味着他在暨
南大学药学院药理学专业的三年硕士学业即将结束。那是在2002年的5月。在《致谢》
部分,李红良说,导师三年来对他的教诲,“令学生终生受益”。
的确,这三年,师生二人的合作可谓“硕果累累”。在这本73页的硕士学位论文中列出
了期间发表的14篇文章(7篇英文,7篇中文)——除去两篇外,李均是第一作者;而其
导师则是绝大多数文章的通讯作者。
作为一名硕士研究生,李的创记录的“高产”引起一片哗然。2001年在其他绝大多数学
生还没有来得及收结果的时候,他就发论文了,而且一发不可收拾,当时就有同学举报
,药学院领导查了,但不了了之。
时光飞逝,当年的硕士“小李”,在获得北京协和医科大学博士,经历2年多国外博士
后,2008年11月从多伦多回到了武汉大学人民医院(当年3月以李红良为第一作者出版
的一篇论文随后被撤稿),并成为了教授。几年下来,李红良组建了据说是100多人的
研究团队,更有“杰青”,“长江”加身,事业可谓如日中天。
然而,2017年是他喜忧参半的一年。喜的是他一年发了5篇《自然医学》论文,忧的是
有举报人向... 阅读全帖 |
|
发帖数: 1 | 34 ◇◇新语丝(www.xys.org)(newxys.com)(xys10.dxiong.com)◇◇
再说饶毅、管坤良实验室联合发表的论文数据造假
·方舟子·
昨天我发出《关于饶毅、管坤良实验室联合发表的论文数据造假的分析》后,
饶毅转给我他当天登在其微信公众号的文章《饶毅、管坤良就论文图片的分析》
(以下简称饶文),作为对“有人匿名散发指饶毅参与署名的9篇论文中图片有
造假问题”的回应。由于第6、7、8、9等四篇文章“不存在需要解释的地方”,
饶文称只需回应第1、2、3、4、5等五篇文章——对此我也同意,我在前文也只
分析了这五篇论文。
但是实际上饶文只回应了“饶毅需要负质量和监管责任的图片”,指的是这
两篇论文中的图片:
论文1:
J Neurosci. 2007 Jan 24; 27(4): 957–968.
p130CAS Is Required for Netrin Signaling and Commissural Axon
Guidance
Guofa Liu, Weiquan Li, Xue Gao,1,2 X... 阅读全帖 |
|
f***y
发帖数: 4447 | 35 https://m.toutiao.com/i6499743832802853390/
1973年,匈牙利数学家 László Fejes Tóth 在 Exploring a Planet 一文中提出了
球带猜想(Zone Conjecture)[1]。该猜想描述了如果一个单位球面被几个球带完全覆
盖,它们的宽度总和至少为 π。44年过去了,以色列理工学院(Technion)的中国数
学家姜子麟和莫斯科物理技术学院(MIPT)的 Alexandr Polyanskii 终于证明了
Fejes Tóth 的猜想,其结果发表于GAFA数学杂志 [2]。他们的证明对于离散几何非常
重要。
○ László Fejes Tóth 猜想。半径为1的单位球体被等宽的区域覆盖。所有区域的
宽度总和的最小值是π。每个区域用不同颜色标记。| 图片来源:MIPT
离散几何学(Discrete Geometry)研究的是点、线、圆、多边形和其他几何对象的组
合性质。例如它会考虑如下问题:在一个球的周围,最多能摆放多少个相同尺寸的球在
它周围?或者,在平面上,如何以最密集的方式排放相同大小的圆?又或者在空间... 阅读全帖 |
|
发帖数: 1 | 36 武汉市地质构造和城市规划建设浅谈
老K世界观
老K世界观
热爱城市规划建设、商业地产和历史的武汉佬
51 人赞同了该文章
地质学和城市规划学是两门专业性极强的学科,如果再结合运用在一个具体的特大城市
进行分析研究,那可以写一本厚书了。
作为一名城建规划的爱好者,看着满城挖、满城建的武汉,总在想:为啥摩天不能建这
里?为啥商务区这样规划?为啥隔壁地铁那么多,我们这还不修?
带着疑问,我参看了1990年至2015年多个高校和机构的相关文献资料,渐渐我越来越了
解了,现在把文献资料的分析结论简单归纳如下,抛砖引玉,如有错误和遗漏,请大神
指出。
武汉地质概况
武汉是一个山水城市,地处华中腹地,位于长江与汉江交汇处,在江汉平原以东,素有
“九省通衢”之称。从土地资源承载力来看,武汉市国土面积仅次于北京、天津,是香
港的八倍。城区地处江汉平原与鄂东南大别山区丘陵、山地的交接地带,地质构造比较
复杂,同时也是一个地壳相对稳定的地区。
中国传统的“风水”和现代的城市规划地质学研究
古人讲究天人合一,在修建房屋之前要“相土尝水”,选择“风水”较好的地段。随着
西方的科学技术的进入,形成了城市规划学和地... 阅读全帖 |
|
b******n
发帖数: 4225 | 37 1. 抗体的选择
对于国内的大多数实验室来讲,做western blot实验选择抗体是个头疼的问题。原因很
简单,买进口抗体捉襟见肘,买国产抗体得需要大无畏的勇气,对于我所在的兰州地区
的实验者而言,感触尤深。在 这五年的western blot实验历程里,我先后用过进口抗
体,进口抗体国内分装包装,国产抗体,质量良莠不齐。
进口抗体一般不会出现闪失:abcam品种全,质量过硬,但价高(3400元/100微升),
而且说是100微升,但至多能吸出来90微升;Sigma的 价最贵;比较有性价比的是CST的
抗体,现在好像是2200元/100微升,我前后用过二十几种, 1:1000的稀释比下,还没
有失过手,100微升通常能完成所有的免疫组化和western blot实验,还有一个优点是
通常量比100微升多出来10微升,唯一的不足是CST的品种实在不多。Santa的多克隆抗
体质量可以,但是选用 Santa的单抗还是有风险,估计这也是业界共识了吧。
进口抗体国内分装包装我也用过不少,呵呵,毕竟是穷人(420元/100微升),大概好
抗体的比例约为50%,如果能做出来,也存在一个问题,就是... 阅读全帖 |
|
i****g
发帖数: 3896 | 38 写得比较有意思,但是不太实用,可能实验条件相差太大。precast gel比自己制的gel
效果要好很多,结果也容易重复。antibody要看运气的,呵呵。
1. 抗体的选择
对于国内的大多数实验室来讲,做western blot实验选择抗体是个头疼的问题。原因很
简单,买进口抗体捉襟见肘,买国产抗体得需要大无畏的勇气,对于我所在的兰州地区
的实验者而言,感触尤深。在 这五年的western blot实验历程里,我先后用过进口抗
体,进口抗体国内分装包装,国产抗体,质量良莠不齐。
进口抗体一般不会出现闪失:abcam品种全,质量过硬,但价高(3400元/100微升),
而且说是100微升,但至多能吸出来90微升;Sigma的 价最贵;比较有性价比的是CST的
抗体,现在好像是2200元/100微升,我前后用过二十几种, 1:1000的稀释比下,还没
有失过手,100微升通常能完成所有的免疫组化和western blot实验,还有一个优点是
通常量比100微升多出来10微升,唯一的不足是CST的品种实在不多。Santa的多克隆抗
体质量可以,但是选用 Santa的单抗还是有风险,估计这... 阅读全帖 |
|
w********h
发帖数: 12367 | 39 【 以下文字转载自 Biology 讨论区 】
发信人: bulletin (bulletin), 信区: Biology
标 题: 穷人的劳斯莱斯——五年western经验谈(转)
发信站: BBS 未名空间站 (Fri Mar 2 23:29:05 2012, 美东)
1. 抗体的选择
对于国内的大多数实验室来讲,做western blot实验选择抗体是个头疼的问题。原因很
简单,买进口抗体捉襟见肘,买国产抗体得需要大无畏的勇气,对于我所在的兰州地区
的实验者而言,感触尤深。在 这五年的western blot实验历程里,我先后用过进口抗
体,进口抗体国内分装包装,国产抗体,质量良莠不齐。
进口抗体一般不会出现闪失:abcam品种全,质量过硬,但价高(3400元/100微升),
而且说是100微升,但至多能吸出来90微升;Sigma的 价最贵;比较有性价比的是CST的
抗体,现在好像是2200元/100微升,我前后用过二十几种, 1:1000的稀释比下,还没
有失过手,100微升通常能完成所有的免疫组化和western blot实验,还有一个优点是
通常量比100微升多出来10... 阅读全帖 |
|
O******e
发帖数: 4845 | 40 ☆─────────────────────────────────────☆
yuuli (听,...听) 于 (Thu Nov 16 23:23:06 2006) 提到:
http://www.cell.com/content/article/fulltext?uid=PIIS0092867406012256&highlight=polymerase
Cell, Vol 127, 317-327, 20 October 2006
Role of the Sigma Factor in Transcription Initiation in the Absence of Core
RNA Polymerase
Hsin-Hsien蔋su,1 Kuei-Min蔆hung,1 Tsung-Ching蔆hen,1 and Ban-Yang蔆hang1,*
1 Institute of Biochemistry, National Chung-Hsing University, Taichung 40227
, Taiwan, Republic of China
从第二个图... 阅读全帖 |
|
t******y
发帖数: 716 | 41 不同蛋白因为侧链的酸碱性,疏水性,电荷性等差异,往往在电泳中跑出来的图形会有
差异,也就是相同的蛋白起条带往往比较类似。这里的图形是指,条带的形状,边缘是
否圆润,像我前面举的例子fig6b,actin的条带表现为底部宽,顶部为圆弧形,而这与
曹文章中其他actin的条带形状相反,其他的actin条带是底部为圆弧形,顶部为宽的直
线。根据蛋白电泳的方向,actin从上往下跑,电泳前沿形成圆弧形似乎更加有道理,
所以,我觉得fig6b的actin图形应该以平行纸面为轴,作180度旋转。 |
|
Z******5
发帖数: 435 | 42 应该不是上样量的问题,我striping后(或者跑的久一点,从45kD处剪开),用beta-
actin杂交,内参的信号已经非常强了,而内源性的c-myc还是很弱。
我用过的两家c-myc的抗体都能杂处很多条带,其中在48-65kD之间有三条条带,转入质
粒过表达显示最上面的条带才是c-myc的(有抗体公司说是表观分子量68kD,有文章说
是64kD,不过所指的是同一条带)。你可以参考这篇文章Nucleolar localization of
hepatic c-Myc: a potential mechanism for c-Myc regulation
关于内源性c-myc杂交很弱的问题,我查过很多文章,多数都是杂带很多,64kD处很弱
。有个别条带很专一,信号很强的,我很怀疑,因为我知道有人拿其它抗体,比如
actin抗体做了个figure,然后说是c-myc杂交的,还发了不错的文章。 |
|
z****i
发帖数: 35 | 43 Caspase-1前体分子量是45kD,剪切后变成20kD的和10kD的蛋白,买的两种抗体能够同
时检测前体和p20,做了很多次都是45kD的位置很多条非特异条带,都分辨不出来目的
条带了,不过条带倒是很浓。20kD的则很淡,基本看不见,把曝光时间增加到很长也还
是很淡,若隐若现的。保证加的刺激能活化Caspase-1。
大家谁这个分子比较有经验,能否指点一下怎么才能跑出来20kD的那条带啊?......用
的是sigma和abcam的抗体。
也试过专门跑小分量的蛋白胶,45kD的带还行,20kD的那条带都不怎么成型了,斑斑点
点的,是不是转膜没转好呢?
谢谢啦! |
|
m***o
发帖数: 272 | 44 western blot上在别的组织上有个短的条带在WT上有,但是在knockout上就消失,别的
非特异条带没有变化。但是我现在觉得这个我找到的shortform不是看到的那个条带,
因为根据这个序列推测蛋白要小很多。
现在我比较确定这个shortform mRNA 的表达在我做的这个组织上很高,因为RT用这个
短的5UTR为forward primer(全长没有这个序列),可以看到很亮的条带,但是别的组
织没有。
我现在也有些不知该怎么往下做,因为抗体不是很好,很多非特异条带。 |
|
F**********6
发帖数: 90 | 45 做了几次CHIP,每次对照IgG都能扩增出很弱的条带(实验组的条带很强!)。请问如
何解决对照IgG有弱条带的问题?
看了些别人发表的CHIP实验数据,绝大部份对照IgG基本上没有条带。但偶然能看到一
两篇文章里的CHIP实验数据里的对照里有弱的条带。
多谢指教 |
|
H****n
发帖数: 26 | 46 最近做一个Transcription Factor(120KD)的ChIP, 很奇怪的事情,--
前面标准的1% formaldyhe RT 10min,0.125M Glycine RT 5 min, PBS wash,
裂解完细胞后,温和的sonication完后,input用western看发现原来条带的位置没有(
或者变的很淡),在下面有两条小的蛋白带。 做了一下RNA pol-II的WB (>150kD <
200kD)也是类似的情况,原来条带的位置淡了很多,在底下出现了两三条size小的条带。
先后试了RIPA (0.1%SDS)和nuclear lysate(1%SDS)裂解,效果一样。只要有
sonication,即使是很温和的5个cycle 30s/30s (该条件下DNA shear都不完全200-
3000bp), 原来蛋白条带就会消失,取而代之的是底下出现的size小的条带。
请问这是sonication 导致大分子量的蛋白容易降解吗?要如何解决? |
|
发帖数: 1 | 47 关于电泳条带的拖尾问题
方先生:
您好!
我是您的超级粉丝,从“基因皇后”事件就开始关注新语丝,一直到现在,
只要打开浏览器,肯定要到新语丝上看一看。
看了7月20号关于韩春雨的文章,不谈其它,我只就电泳条带的拖尾现象谈
一下我的认识。
一般情况下,电泳条带拖尾状况正如方先生所说,边缘拖后,中间超前。我
原先也是一直是这样认为的,但是正是这样的认识,差点冤枉了一个研究生。有
一年研究生预答辩时,我看到同组老师的一个研究生ppt中一张蛋白电泳图非常
特别,蛋白条带中间拖后,边缘超前。我就问学生为什么会这样?这个学生说他
也不知道为什么,跑出来的结果就是这样的。maker也是这样。我非常怀疑,但
是保险起见,我没有再争论下去,而是会后自己查了一下,终于发现了原因。他
的蛋白电泳分析的是超低分子量蛋白,用的是Tricine-SDS-PAGE,在这种条件下,
加上电泳仪电压不均衡等其它原因,小分子量蛋白的确会出现边缘超前,中间拖
后的结果。请看网上的一些图片:
http://www.chem17.com/offer_sale/detail/11291438.... 阅读全帖 |
|
r**a
发帖数: 121 | 48 我们实验室用的一个主要的抗体
是以杂交瘤细胞上清的形式存放在-80度冰箱
存放了15年了(95年的)
现在情况是
western在细胞上不怎么好
大部分时候,杂带多,在目的条带处也有3个条带
有可能是磷酸化,但是每次三个条带之间的分布不同
但如果进行组织样品的western
有时能得到一个还可以的条带
稀释倍数从1:20到现在1:5
所以,我想复苏细胞重新弄点抗体
但是,老板和一个资深博士都说
抗体这样可以保存很长时间,绝对没有问题的
(因为我们这样的抗体在-80度还有很多保存着)
所以,询问一下这里的前辈
是不是真的这样保存,抗体很稳定? |
|
f*********r
发帖数: 1233 | 49 与细胞/组织匀浆不同,血清中dna极低。粘稠主要因为蛋白和血脂。楼上说超高速离心
,是个不错的办法。能够去掉很多悬浊微粒。如果样品量有限,可以加过lysis buffer
以后再离心。
稀释总蛋白浓度。如果5ul/well这个量不能再降低,可以象楼上所说,用大胶、厚胶,
想办法增加volume/well。
如果有条件,可以用filter过滤掉不需要的蛋白。一般,条带扭曲多发生在小分子量。我估计你要的蛋白就不大。那么,你可以根据分子量,过滤掉大分子蛋白。millipore就有很多不同孔径的过滤系统可供选择。
条带形状不规整,很大程度上还因为蛋白denature得不够好。可以适当在loading buffer里增加sds浓度。
由于loading buffer与running buffer之间在表面活性上有差异,可以造成条带的扭曲
。为此,可以选用taurin buffer(代替glycin buffer)以弱化条带扭曲。甚至,有的
pi曾经建议,在上样之前,空跑至少10分钟。
用一般的样品跑胶,那么胶的品牌与型号之间的优劣不是很明显。你的sample比较特殊
,可能就放大了各厂家产品之间 |
|
{kind=link}